差异基因富集分析(R语言——GO&KEGG&GSEA)
创作时间:
作者:
@小白创作中心
差异基因富集分析(R语言——GO&KEGG&GSEA)
引用
CSDN
1.
https://blog.csdn.net/m0_74823094/article/details/144343797
在基因表达数据分析中,获得差异基因集后,进行通路富集分析是理解这些基因功能和生物学意义的重要步骤。本文将详细介绍如何使用R语言进行GO(Gene Ontology)、KEGG(Kyoto Encyclopedia of Genes and Genomes)和GSEA(Gene Set Enrichment Analysis)富集分析,帮助研究人员深入挖掘差异基因的生物学意义。
1. 准备差异基因集
在进行通路富集分析之前,我们需要准备差异基因集。通常,我们会将差异基因分为上调基因和下调基因分别进行分析。以下代码展示了如何从上次分析中获取差异基因集:
##载入所需R包
library(readxl)
library(DOSE)
library(org.Hs.eg.db)
library(topGO)
library(pathview)
library(ggplot2)
library(GSEABase)
library(limma)
library(clusterProfiler)
library(enrichplot)
##edger
edger_diff <- diff_gene_Group
edger_diff_up <- rownames(edger_diff[which(edger_diff$logFC > 0.584962501),])
edger_diff_down <- rownames(edger_diff[which(edger_diff$logFC < -0.584962501),])
##deseq2
deseq2_diff <- diff_gene_Group2
deseq2_diff_up <- rownames(deseq2_diff[which(deseq2_diff$log2FoldChange > 0.584962501),])
deseq2_diff_down <- rownames(deseq2_diff[which(deseq2_diff$log2FoldChange < -0.584962501),])
##将差异基因集保存为一个list
gene_diff_edger_deseq2 <- list()
gene_diff_edger_deseq2[["edger_diff_up"]] <- edger_diff_up
gene_diff_edger_deseq2[["edger_diff_down"]] <- edger_diff_down
gene_diff_edger_deseq2[["deseq2_diff_up"]] <- deseq2_diff_up
gene_diff_edger_deseq2[["deseq2_diff_down"]] <- deseq2_diff_down
2. 进行通路富集分析
接下来,我们将进行GO和KEGG富集分析。这里使用循环处理上调和下调基因集,以提高效率。
for (i in 1:length(gene_diff_edger_deseq2)){
keytypes(org.Hs.eg.db)
entrezid_all = mapIds(x = org.Hs.eg.db,
keys = gene_diff_edger_deseq2[[i]],
keytype = "SYMBOL", #输入数据的类型
column = "ENTREZID")#输出数据的类型
entrezid_all = na.omit(entrezid_all) #na省略entrezid_all中不是一一对应的数据情况
entrezid_all = data.frame(entrezid_all)
##GO富集##
GO_enrich = enrichGO(gene = entrezid_all[,1],
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID", #输入数据的类型
ont = "ALL", #可以输入CC/MF/BP/ALL
#universe = 背景数据集我没用到它。
pvalueCutoff = 1,qvalueCutoff = 1, #表示筛选的阈值,阈值设置太严格可导致筛选不到基因。可指定 1 以输出全部
readable = T) #是否将基因ID映射到基因名称。
GO_enrich_data = data.frame(GO_enrich)
write.csv(GO_enrich_data,paste('GO_enrich_',names(gene_diff_edger_deseq2)[i], '.csv', sep = ""))
GO_enrich_data <- GO_enrich_data[which(GO_enrich_data$p.adjust < 0.05),]
write.csv(GO_enrich_data,paste('GO_enrich_',names(gene_diff_edger_deseq2)[i], '_filter.csv', sep = ""))
###KEGG富集分析###
KEGG_enrich = enrichKEGG(gene = entrezid_all[,1], #即待富集的基因列表
keyType = "kegg",
pAdjustMethod = 'fdr', #指定p值校正方法
organism= "human", #hsa,可根据你自己要研究的物种更改,可在https://www.kegg.jp/brite/br08611中寻找
qvalueCutoff = 1, #指定 p 值阈值(可指定 1 以输出全部)
pvalueCutoff=1) #指定 q 值阈值(可指定 1 以输出全部)
KEGG_enrich_data = data.frame(KEGG_enrich)
write.csv(KEGG_enrich_data, paste('KEGG_enrich_',names(gene_diff_edger_deseq2)[i], '.csv', sep = ""))
KEGG_enrich_data <- KEGG_enrich_data[which(KEGG_enrich_data$p.adjust < 0.05),]
write.csv(KEGG_enrich_data, paste('KEGG_enrich_',names(gene_diff_edger_deseq2)[i], '_filter.csv', sep = ""))
}
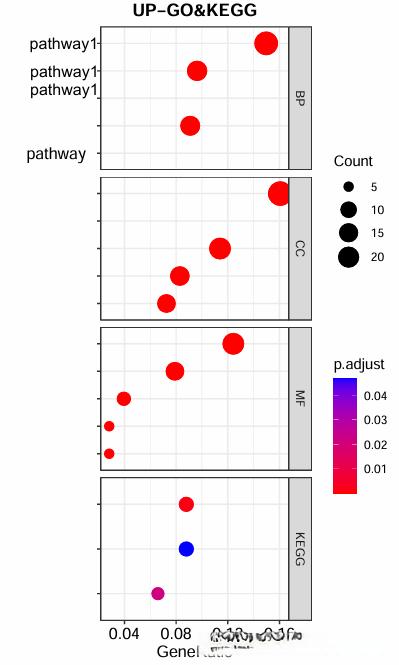
3. 通路富集情况可视化
富集分析的结果需要通过可视化的方式展示,以便更好地理解数据。这里介绍一种简单的气泡图绘制方法。
##GO&KEGG富集BPCCMFKEGG分面绘图需要分开处理一下,富集结果里的ONTOLOGYL列修改
GO_enrich_data_BP <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "BP")
GO_enrich_data_CC <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "CC")
GO_enrich_data_MF <- subset(GO_enrich_data, subset = GO_enrich_data$ONTOLOGY == "MF")
##提取GO富集BPCCMF的top5
GO_enrich_data_filter <- rbind(GO_enrich_data_BP[1:5,], GO_enrich_data_CC[1:5,], GO_enrich_data_MF[1:5,])
##重新整合进富集结果
GO_enrich@result <- GO_enrich_data_filter
##处理KEGG富集结果
KEGG_enrich@result <- KEGG_enrich_data
ncol(KEGG_enrich@result)
KEGG_enrich@result$ONTOLOGY <- "KEGG"
KEGG_enrich@result <- KEGG_enrich@result[,c(10,1:9)]
##整合GO KEGG富集结果
ego_GO_KEGG <- GO_enrich
ego_GO_KEGG@result <- rbind(ego_GO_KEGG@result, KEGG_enrich@result[1:5,])
ego_GO_KEGG@result$ONTOLOGY <- factor(ego_GO_KEGG@result$ONTOLOGY, levels = c("BP", "CC", "MF","KEGG"))##规定分组顺序
##简单画图
pdf("edger_diff_up_dotplot.pdf", width = 7, height = 7)
dotplot(ego_GO_KEGG, split = "ONTOLOGY", title="UP-GO&KEGG", label_format = 60, color = "pvalue") +
facet_grid(ONTOLOGY~., scale = "free_y")+
theme(plot.title = element_text(hjust = 0.5, size = 13, face = "bold"), axis.text.x = element_text(angle = 90, hjust = 1))
dev.off()
4. GSEA富集分析
GSEA是一种基于基因集的富集分析方法,能够更全面地评估基因集在差异表达中的富集情况。以下是GSEA分析的代码示例:
##GSEA官方网站下载背景gmt文件并读入
geneset <- list()
geneset[["c2_cp"]] <- read.gmt("c2.cp.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_kegg_legacy"]] <- read.gmt("c2.cp.kegg_legacy.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_kegg_medicus"]] <- read.gmt("c2.cp.kegg_medicus.v2023.2.Hs.symbols.gmt")
geneset[["c2_cp_reactome"]] <- read.gmt("c2.cp.reactome.v2023.2.Hs.symbols.gmt")
geneset[["c3_tft"]] <- read.gmt("c3.tft.v2023.2.Hs.symbols.gmt")
geneset[["c4_cm"]] <- read.gmt("c4.cm.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_bp"]] <- read.gmt("c5.go.bp.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_cc"]] <- read.gmt("c5.go.cc.v2023.2.Hs.symbols.gmt")
geneset[["c5_go_mf"]] <- read.gmt("c5.go.mf.v2023.2.Hs.symbols.gmt")
geneset[["c6"]] <- read.gmt("c6.all.v2023.2.Hs.symbols.gmt")
geneset[["c7"]] <- read.gmt("c7.all.v2023.2.Hs.symbols.gmt")
##进行GSEA富集分析,这里也是写了个循环
gsea_results <- list()
for (i in names(gene_diff)){
geneList <- gene_diff[[i]]$logFC
names(geneList) <- toupper(rownames(gene_diff[[i]]))
geneList <- sort(geneList,decreasing = T)
for (j in names(geneset)){
listnames <- paste(i,j,sep = "_")
gsea_results[[listnames]] <- GSEA(geneList = geneList,
TERM2GENE = geneset[[j]],
verbose = F,
pvalueCutoff = 0.1,
pAdjustMethod = "none",
eps=0)
}
}
##批量绘图,注意这里如果有空富集通路,会报错!
for (j in 1:nrow(gsea_results[[i]]@result)) {
p <- gseaplot2(x=gsea_results[[i]],geneSetID=gsea_results[[i]]@result$ID[j], title =
gsea_results[[i]]@result$ID[j])
pdf(paste(paste(names(gsea_results)[i], gsea_results[[i]]@result$ID[j], sep =
"_"),".pdf",sep = ""))
print(p)
dev.off()
}
5. GSEA富集分析结果
GSEA富集分析的结果可以通过热图或条形图等方式展示。以下是GSEA富集分析的简单图形示例:
通过以上步骤,我们可以系统地完成差异基因的通路富集分析,并通过可视化方式展示分析结果。希望这篇文章能对大家的科研工作有所帮助。
热门推荐
剃须刀选购攻略
贷款无力偿还债务新政策怎么执行?
克格勃秘密实验室:特异功能与心理操控
时点数的定义与计算方法:在数据分析中的重要应用
成都轨道交通30号线一期、13号线一期最新进展来了!
南京大学现代工程与应用科学学院微纳光学研究进展
铜价飙涨!幕后是谁?地缘政治冲突加剧供应端扰动
你的电动自行车换新了吗?
预计11月底满足通航条件!新工艺、数字化、智能化助力昆明长水国际机场新跑道建设
西交利物浦大学4+0国际本科好吗?认可度咋样?
瑜伽最初的起源和目的
瑜伽最初的起源和目的
华东地区首例!华山医院成功开展腹腔镜辅助成人全肝移植手术
翰墨飘香 医道远行 北京清华长庚医院肝肾移植进修之学术之旅
精选内容集|大英博物馆探秘之旅 感受千年文明 醉心艺术与历史的交融
美国名校云集的四大州:从教育资源到生活指南
STM32 60秒倒计时代码详解(适合初学者) 完整代码+仿真图
资产等于权益的原理是什么?这一原理在财务管理中有哪些应用?
八字有多少种组合,一共有多少种可能的八字?
八字合在命理中代表的含义与重要性 理解八字相合的深层次意义
《暗黑破坏神不朽》猎魔人套装推荐 猎魔人憎恨普攻流派攻略
敏感性肌肤适合的面膜
《冰心诀》全文仅 114 字 解锁心灵宁静密码,从容拥抱生活。
中药肾气丸治疗肝损伤新突破:浙中医团队揭示其作用机制和活性成分
哲学探索-无为而治老子智慧的现代解读
Telegram两种账户类型详解:普通用户账号 vs 企业账号
万物皆可“沉浸” 身临其境“触摸”济南城
打工人自救放松之5个舒缓手腕酸痛的实用小方法
欧亨利式结尾是什么(浅谈“欧·亨利式结尾”及其文学影响)
多久可查艾滋?快速了解艾滋病毒检测时间窗