腰椎间盘突出重吸收机制
腰椎间盘突出重吸收机制
腰椎间盘突出是一种常见的脊柱疾病,其治疗方式多样,包括手术和保守治疗。近年来,研究发现部分患者的突出髓核可以自发性重吸收,这一现象为保守治疗提供了新的可能性。本文将从多个角度探讨腰椎间盘突出自发性重吸收的机制,帮助读者更好地理解这一现象。
腰椎间盘突出不手术也能好,这就涉及到一个概念:「腰椎间盘突出自发性重吸收」,它是指椎间盘突出症(lumbar disc herniation,LDH)患者未经侵入性治疗而发生的突出髓核自发性消失或明显缩小。
目前相关文献报道多为病例报道或回顾性研究。回顾文献,最经典的腰椎间盘自发性重吸收的病例当是 2016 年发表在新英格兰医学杂志(NEJM)的病例。
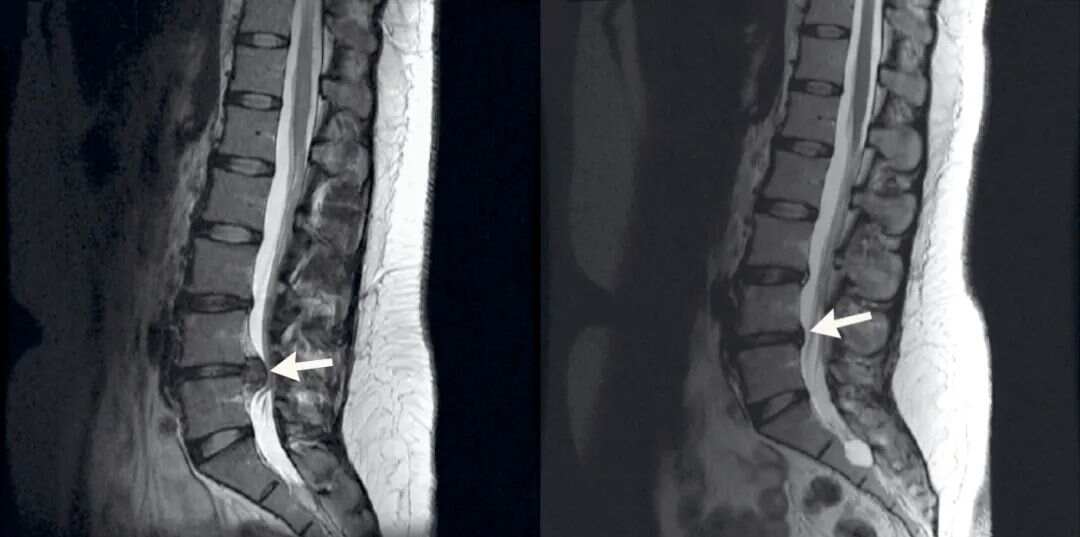
图一:LDH 患者 L4/5 在出现症状 5 个月后发生自发性消退
患者女,29 岁,因「右小腿疼痛并感觉异常」就诊,大小便正常。起始腰椎磁共振提示腰 L4/5 突出并椎管狭窄,神经根压迫严重(左图)。患者选择保守治疗,5 个月后复查 MRI(右图)发现突出的椎间盘自发性吸收了,并且临床症状基本缓解。
图二:LDH 患者 L4/5 发生自发性消退,MRI 显示 3 年后在 L5/S1 上出现新的突出。图源:DOI: 10.1155/2016/1538072
图三:LDH 患者 MRI 显示重吸收的时间为出现症状后 4 个月,图源:DOI: 10.1016/j.neuchi.2018.12.001
以上影像学研究表明,部分突出髓核可随着时间自然消退,这为 LDH 后在未出现严重的神经症状的情况下采取保守治疗提供了客观依据,同时也对手术治疗提出了更严格的适应证。
目前,研究已证实 LDH 后髓核可以部分或全部重吸收,髓核自发性重吸收时间一般为 2~12 个月,且后纵韧带破裂型间盘突出的髓核重吸收率高于未破裂型,但概率很低,文献报道证据等级低,需慎重看待。
目前,关于突出髓核的自发性重吸收机制尚未完全阐明,本文即从以下 8 个机制阐述 LDH 髓核自发性重吸收的机制。
一、力学机制
著名生物力学专家 Stuart McGill 教授在《腰部疾患: 循证预防与康复》提出:
不良的坐位和立位姿势、腰椎过度机械负荷、频繁的躯干运动、反复的腰椎屈曲和扭转运动均可导致 LDH。
在生物力学与机械力学的影响下,椎间盘的核心受压会引起软骨终板向椎体内膨出,导致星状骨折;纤维环在受压时会使外层的纤维环向外膨胀,内层的纤维环向内膨胀,致使纤维环分层或脱层,而髓核组织会沿着终板骨折处或纤维环破裂处突出,从而形成 Schorml 结节和椎间盘突出。
箭头所指即为 L3 椎体的 Schmorl 结节
Schorml 结节是指椎间盘髓核组织经破裂的软骨终板突入邻近的椎体松质骨内。这一经典的理论由德国医生 Schmorl 于 1927 年首先提出, 一直被绝大多数脊柱外科医生所接受。
由于上述异常应力和剪切力的作用,纤维环破裂髓核突出会改变椎间盘力学特性,而机械力学也进一步通过改变细胞内稳态来调控炎症因子、生长因子、金属基质蛋白酶等相关因子的表达,导致腰椎间盘退变。
所以,减轻腰椎机械压力可以减轻 LDH,并在一定程度上促进髓核的重吸收。
二、炎症免疫反应机制
腰椎间盘是人体最大的无血管器官,髓核自形成以来就被周围的纤维环和终板围绕,这种独特的结构将髓核从机体的免疫系统中分离出来,即为「血液-髓核屏障」(blood-nucleus pulpo-sus barrier,BNB)。
髓核组织中的蛋白多糖和胶原蛋白具有强自身免疫原性,当髓核与血液循环接触后,在巨噬细胞或其他抗原提呈细胞的识别、处理和呈递作用下会激活 T 或 B 淋巴细胞。
活化的 T 细胞:可产生免疫调节细胞和效应细胞,通过延迟型超敏反应或细胞毒性反应破坏椎间盘组织;
活化的 B 细胞:可分化为浆细胞分泌抗体,这些抗体通过巨噬细胞的吞噬和补体系统用自身抗原破坏髓核组织。
自身免疫反应:可通过细胞免疫与体液免疫破坏或溶解髓核细胞,除此之外还可诱导单核巨噬细胞等炎性细胞浸润,分泌 IL-1β、IL-6、IL-17、TNF-α 等炎症因子,炎症因子虽然不能直接降解突出的髓核,但可以通过促进降解酶,如 MMP 和金属蛋白酶域蛋白(a disintegrin and metalloproteinase with thrombospondin motifs,ADAMTS),减少蛋白多糖和胶原蛋白的表达,降解髓核细胞外基质,促进突出髓核重吸收。
Li 等研究发现,TNF-α 可以通过抑制髓核 G1 期细胞周期来阻滞髓核细胞的生长,并加速髓核细胞凋亡。Robert 等研究表明,炎症可引起髓核细胞的生物力学和结构变化,通过调节水的渗透性来调控髓核细胞大小。
炎症因子还可进一步诱导椎间盘细胞产生趋化因子,如人巨噬细胞趋化蛋白-1(human macrophage chemoattractant protein-1,MCP-1),进一步募集巨噬细胞,从而形成炎症级联反应。
①巨噬细胞可主动吞噬突出髓核,并将其在充满胶原降解酶的溶酶体中进行处理;
②巨噬细胞可将分泌的溶酶体酶送至细胞外分解椎间盘基质。
Henson 等对 LDH 患者随访 1 年后发现,巨噬细胞浸润数量与突出到体循环的髓核大小呈正相关,突出的髓核越大越易被重吸收。
最新的研究发现,巨噬细胞分为有促炎作用的 M1 型、抗炎作用的 M2a 型和具有重塑功能的 M2c 型。Kenneth 等在退变性腰椎间盘标本中均发现了 M1 表型和 M2c 表型的巨噬细胞浸润增多,表明退变的椎间盘处于促炎和重构状态。
所以,当纤维环破裂后突出髓核引起自身免疫反应,并与免疫细胞及炎症因子相互作用,促进突出的髓核重吸收。
三、新生血管作用机制
椎间盘是人体内最大的无血管组织,营养物质通过软骨终板及纤维环渗透到髓核细胞,满足髓核的代谢需求。
突入硬膜外腔的椎间盘组织最具特征性的变化是,椎间盘组织表面新生大量毛细血管和薄壁血管,新生血管的长入有助于巨噬细胞的浸润,通过吞噬作用可使突出的髓核组织缩小甚至消失。
Silva 等在退变性腰椎间盘或在加入炎症因子培养的腰椎间盘研究中发现,巨噬细胞会分泌出大量的血管内皮生长因子(vascular endothelial growth factor,VEGF),提示巨噬细胞可通过 VEGF 介导椎间盘内新生血管形成与聚集。次年,有学者研究发现突出髓核组织在 VEGF 的作用下可促进新生血管长入髓核。
其他生长因子,如基质细胞衍生因子 1(stromal cell derived factor 1,SDF1)也可以促进腰椎间盘新生血管的形成;如血小板源生长因子(platelet derived growth factor,PDGF)和转化生长因子 β1(transforming growth fac-tor β1,TGF-β1)也在突出髓核组织中表达增高,二者均可促进新生血管生成,并与炎症因子 TNF-α 和 IL-1 相互作用,通过诱导巨噬细胞和提高 MMPs 的表达来促进突出髓核重的吸收。
所以,新生血管与炎症因子相互作用,共同促进突出髓核重吸收。
四、外泌体
外泌体是近年来发现的一种新型细胞通讯机制,外泌体中包含多种细胞因子、信号蛋白、脂质,以及调节性核酸等成分外泌体膜与靶细胞膜融合后将信息物质释放入靶细胞,从而传递并调节信息的表达。
外泌体介导的细胞-细胞通路是目前生物和医学研究中发展最迅速的领域之一,目前已表明人体内许多类型的细胞都可以释放外泌体。
研究发现,间充质干细胞(mesenchymal stromal cells,MSCs)主要是通过其分泌的外泌体经旁分泌途径在 MSCs 与靶标细胞间发挥其通讯功能。
MSCs 来源的外泌体可以诱导 Th1 细胞向 Th2 细胞转变,从而分泌一系列炎症因子如 IL-4、IL-5Ra 和 IL-13 等参与机体的免疫反应。此外,在纤维环培养基中加入内皮细胞分泌的外泌体,可以促进 MMPs 的表达,而 MMPs 可以进一步降解髓核细胞外基质。
所以,外泌体可能通过介导炎症因子释放和提高 MMPs 的表达参与髓核重吸收放和提高 MMPs 的表达参与髓核重吸收。
五、细胞凋亡机制
细胞凋亡是细胞通过基因及其产物调控而发生的一种程序性细胞死亡,细胞凋亡在髓核重吸收中发挥一定作用,LDH 患者椎间盘细胞凋亡因子的表达明显高于健康人,且凋亡相关因子可以介导髓核细胞凋亡。
研究发现,髓核组织中高表达的 IL-1β 可以降低 Bcl-2/Bax 比值,增强促凋亡因子细胞色素 C(cytochrome C,CytC)的释放,CytC 与凋亡蛋白酶活化因子(apoptotic protease activating factor 1,Apaf-1)和 Caspase-9 结合形成「凋亡体」,进一步激活 Caspase3 并最终引起细胞凋亡 。
此外有研究报道,参与椎间盘退变的活性氧(reactive oxygen species,ROS)可通过激活 NF-kB 通路导致髓核细胞凋亡。
凋亡相关因子(factor related apoptosis,FAS)与 FAS 配体(factor related apoptosis ligand,FASL)结合,也可以启动凋亡信号的转导引起细胞凋亡。
有学者发现退行性椎间盘中髓核 FASL 的表达明显低于健康椎间盘,纤维环破裂后,突出髓核引起的免疫反应及沿新生血管浸润的细胞毒性 T 淋巴细胞和 NK 细胞被活化并表达 FASL,与髓核表面的 FAS 结合,诱导髓核细胞凋亡。
同时,研究发现 TNF-α 还可激活 FAS/FASL 途径介导髓核细胞凋亡,即髓核细胞凋亡可导致髓核细胞数量减少,进而影响胶原蛋白、蛋白聚糖等椎间盘髓核组织的细胞外基质合成,从而达到促进髓核重吸收的作用。
六、细胞外基质合成与分解代谢失调机制
髓核由髓核细胞和细胞外基质组成(extracellular matrix,ECM),髓核细胞外基质主要由 Ⅱ 型胶原蛋白和蛋白多糖组成,可由 MMPs 诱导降解。细胞外基质合成与降解由细胞外基质修饰酶如 MMPs、ADAMTS 和组织金属蛋白酶抑制剂(Tissue in-hibitor of metalloproteinases,TIMPs)调控。
有学者研究发现,退变椎间盘标本中,ADAMTS-4 和 ADAMTS-5 的表达显著增加,二者是椎间盘蛋白多糖降解的必须因子。
同时,研究报道 IL-1β 和 TNF-α 可增加髓核细胞中 MMP-3、ADAMTS-4/5 的生成,且 MMPs-3 是降解髓核细胞外基质的关键酶。
此外,研究发现 MMPs 的拮抗剂 TIMPs 在退变的腰椎间盘髓核细胞中表达下降,抑制细胞外基质合成。
所以,髓核突出后基质降解酶(如 MMPs 或 ADAMTS)在髓核细胞中表达增高,二者可通过降解髓核细胞外基质蛋白多糖和Ⅱ型胶原蛋白达到促进突出髓核重吸收的作用。
七、自噬机制
自噬是一个重要的分解代谢过程,可以清除受损的细胞器和蛋白质,在许多疾病中参与细胞的凋亡和衰老。成熟的自噬体可与溶酶体融合形成自噬溶酶体,衰老或受损的细胞质在溶酶体相关膜蛋白(lysosomal associated membrane protein,LAMP)的帮助下被降解。
过度自噬可以介导髓核细胞死亡。
Gruber 等通过体内基因分析发现,退化的椎间盘中自噬相关基因表达高于健康的椎间盘。同时,有学者对于破裂型与未破裂型髓核组织的对比发现,两者类型均有自噬现象的发生,且破裂型髓核细胞中的自噬活动远高于未破裂型。
此外,有学者发现大鼠髓核细胞在压力作用下也可以通过自噬作用介导髓核细胞死亡。
通过上述研究,发现髓核突出后会诱导自噬反应介导髓核细胞死亡,这有利于髓核的重吸收。
八、脱水作用机制
髓核内的蛋白多糖具有较高的阴离子电荷,可吸引并保留大量水分子,所以髓核细胞外基质约 80% 是水。
髓核突破后纵韧带接触水溶液后,因自身高渗透性可使其进一步吸水膨胀,使突出的髓核体积增大,随着突出髓核细胞外基质蛋白多糖降解,髓核细胞高渗性下降,水分流失,突出髓核的体积会随之变小。
影像学突出髓核 MRI T2 值会降低,这种变化证实,突出髓核会发生脱水,且突出的髓核体积会随时间的迁移逐渐缩小。
突出髓核还通过降低水分子转运减小突出髓核体积。
研究发现,腰椎间盘的膜运输可能受水通道蛋白家族(aquaporin,AQP)的调控。在椎间盘中发挥促进水稳态、溶质转运、调节细胞体积等重要作用。
此外,有研究证明 AQP1 和 AQP3 表达于髓核细胞,参与调控髓核细胞的水含量。
Robert 等发现炎症可导致髓核中 AQP1 表达下降,从而使髓核含水量下降,这可能是造成突出髓核体积缩小的原因之一。
曹国永等则发现,AQP3 在退变椎间盘中的表达降低,从而导致椎间盘软骨终板渗透作用异常,髓核细胞代谢产物过度堆积,破坏椎间盘髓核细胞的代谢环境,加速髓核细胞的凋亡。
所以,髓核突出后会因渗透压的下降而脱水,并在水通道的调控下进一步减少细胞外水分子进入髓核细胞内,从而使突出的髓核缩小。
总 结
随着重吸收现象临床报道不断增多,相关的基础研究也不断深入,关于重吸收的机理逐渐明朗,人们逐渐认识到手术并非腰椎间盘突出症最终的更不是唯一的治疗手段,在没有出现严重神经损伤时,重吸收现象的发现为保守治疗 LDH 提供了更多的可能性。
但是,我们也要认识到重吸收的发生并非常见现象,即使已经证实破裂型突出更易于发生重吸收,但这种情况下发生重吸收也不是必然,重吸收还与其他多种因素相关,如研究方法、纳入标准、随访时间等。目前, 已有相关研究根据 LDH 重吸收的好发因素进行预测,但结果不一。
预测是否能发生重吸收,应进行综合评估。
当经过一定时间严格的保守治疗后,患者症状及影像学均未有明显改观,则此时手术治疗也许是需要积极考虑的一个选择。